屠强研究组开发Decode-seq方法显著提高差异表达基因分析的准确性
| 屠强研究组开发Decode-seq方法显著提高差异表达基因分析的准确性 | ||
|
鉴定差异表达基因是许多生物医学研究项目的基础步骤,利用转录组进行差异表达(Differential Expression, DE)分析是目前最主流的方法,得到了广泛应用。例如,两个常用于转录组DE分析的算法 edgeR 和 DESeq2 已经被引用了超过上万次。
在DE分析中,如果使用的生物学重复数不足,会影响到结果的准确性,生物统计学家们长期以来呼吁要提高重复数。然而,由于传统转录组方法的实验流程较为复杂昂贵,大多数研究仅使用 2-3 对生物学重复。使用这么少重复数的分析,敏感性(sensitivity)较低,只有差异表达最显著的基因才能被鉴定出来;错误发现率(FDR)也很高,结果中存在大量的假阳性。因此,需要一种新的实验方法,有效解决生物学重复不足的问题,提高DE分析的准确性。
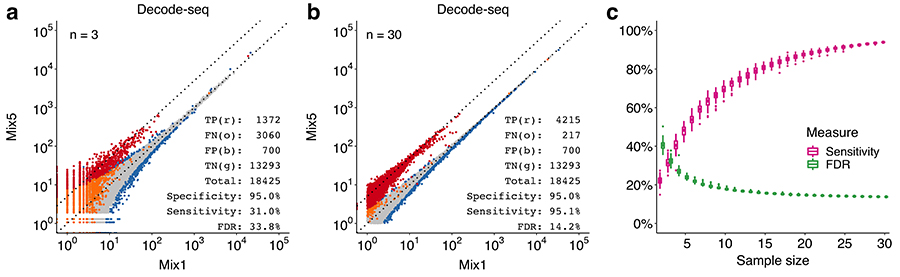
分子发育生物学国家重点实验室屠强研究组开发了一种新的转录组实验方法,称为Decode-seq。该方法利用多种分子标签,可以在一个测序文库中同时检测数十个样品,大幅降低了建库成本;富集转录本5’端测序,相比全长测序降低了测序成本,也避免了3‘端测序造成的困难;文库结构兼容通用测序流程,不需要定制参数,可以与任意标准文库同时上机,因此对测序量要求也非常灵活。与传统转录组方法相比,Decode-seq的成本降低到约10%,非常适用于大规模的DE分析。在验证实验中,使用30对重复的DE分析结果相对于3对重复,敏感性从31% 提高到 95%,错误发现率从 34% 降低到 14%,显著改善了结果的准确性。Decode-seq 方法还可以应用于微量样品,例如对青鳉鱼发育早期的雌雄性腺进行多达30对重复的转录组分析,鉴定出了多个雌雄差异表达基因,其中一些被证明对生殖细胞的正常发育是必须的。
综合这些结果可知,大幅增加生物学重复可以显著提高差异表达基因鉴定的准确性,而Decode-seq 为此提供了一个可行的实验方法。因此,除非因样品珍贵、需要研究转录本全长等原因,否则应避免仅使用2-3对生物学重复的转录组分析策略。Decode-seq 方法的实验流程和分析程序都已经在研究组主页上提供下载。
该成果于2020年3月23日正式发表于 Genome Biology 杂志(DOI:10.1186/s13059-020-01966-9),文章标题为“Decode-seq: a practical approach to improve differential gene expression analysis”。屠强研究组博士研究生李莹姝为该论文的第一作者,屠强研究员为该论文的通讯作者。该研究得到国家自然基金和中科院先导项目的资助。
|
附件下载: